By going through these CBSE Class 12 Chemistry Notes Chapter 9 Coordination Compounds, students can recall all the concepts quickly.
Coordination Compounds Notes Class 12 Chemistry Chapter 9
Coordination Compounds or complex compounds are usually formed by the transition metals ¡n which the metal atoms or ions are bound to a number of anions or neutral molecules Chlorophyll, haemoglobin and vitamin B12 are coordination compounds of magnesium, iron and cobalt respectively.
Wernet’s Theory of Coordination Compounds:
Alfred Werner, a Swiss chemist proposed the concept of a primary valence and a secondary valence for a metal ion. Werner in 1998 propounded his theory of coordination compounds.
The main postulates are:
- In coordination compounds metals show two types of linkages (valences) primary and secondary.
- The primary valences are normally ionisable and are satisfied by negative ions.
- The secondary valences are non-ionizable. These are satisfied by neutral molecules or negative ions. The secondary valence is equal to the coordination number and is fixed for a metal.
- The ion.s/groups bound by the secondary linkages to the metal have characteristic spatial arrangements corresponding to different numbers.
Such spatial arrangement is called coordination polyhedra. The species within the square bracket are coordination entities or complex part and the ions outside the coordination entities or complex part are called counter ions.
Werner further postulated that octahedral, tetrahedral, and square planar geometrical shapes are more common in coordination compounds of transition metal.
[CO(NH3)6]3+, [Cr(H2O)6]3+, [Cr Cl (NH3)5]2+, [COCl2(NH3)4]+ are octahedral complexes [NiCO)4] and [PtCl4]2- are tetrahedral and square planar respectively.
Difference between a double salt and a complex: Both double salts, as well as coordination compounds, are formed by the combination of two or more stable compounds in a stoichiometric ratio.
- KCl + MgCl2 + 6 H2O → KCl. MgCl2. 6H20 (carnallite)
- FeSO4 + (NH4)2SO4 + 6 H2O → FeSO4. (NH4)2 SO4. 6H2O (Mohr’s salt)
- 4 KCN + Fe (CN)2 → K4[Fe(CN)6]
- CoCl3 + 6 NH3 → [Co(NH3)6]Cl3
1 and 2 are examples of double salts
3 and 4 are examples of coordination compounds.
Double salts lose their identity in aqueous solutions. They dissociate into simple ions completely when dissolved in water.
Coordinate complexes retain their identity both in the solid-state and in aqueous solutions. For example [Fe (CN)6]4- does not dissociate into Fe2+ and CN– ions.
→ Coordination Sphere: The central atom/ion and the ligands attached to it are enclosed in a square bracket is collectively called the Coordination sphere. For example in K4[Fe (CN)6], [Fe(CN)6]4- is coordination sphere and ionisable group K+ written outside the coordination group is called Counterion.
→ Coordination Polyhedron: The spatial arrangement of the ligand atoms which are directly attached to the central atom/ion defines polyhedron about the central atom/ion [Co(NH3)6]3+, [Ni (CO)4] and [Pt Cl]2- are respectively octahedral, tetrahedral and square planar coordination polyhedra.
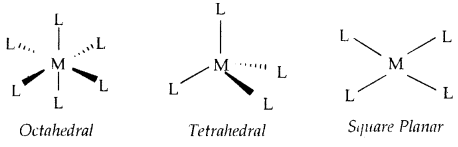
(Shapes of different coordination polyhedra. M = Central metal/ ion and L = a unidentate ligand)
→ Oxidation Number of Central Atom: The oxidation number of the central atom in a complex is defined as the charge it would carry if all the ligands are removed along with the electron pairs that are shared with the central atom. Oxidation no. of copper in [Cu (CN)4]3- is + 1 and it is written as Cu (I).
→ Coordination Entity/Complexion: A coordination entity constitutes a central metal atom or ion bonded to a fixed number of ions or molecules. For example, [CoCl (NH3)3] is a coordination entity in which the cobalt ion is surrounded by three ammonia molecules and three chloride ions. Other examples are [Ni (CO)4], [PtCl, (NH3)2], [Fe (CN)6]3-, [CO(NH3)6]3+.
→ Central atom/ion: In a coordination entity, the atom/ion to which a fixed number of ions/ groups are bound in a definite geometrical arrangement around it, is called the central atom or ion. For example, the central atom/ion in the coordination entities: [NiCl2 (H2O)4], [COCl(NH3)5]2+, [Fe(CN)6]3- are Ni2+, CO3+ and Fe3+, respectively. These central atoms/ions are also referred to as Lewis acids.
→ Ligands: The ions or molecules bound to the central atom/ion in the coordination entity are called ligands. These may be simple ions such as Cl, small molecules such as H2O or NH3, larger molecules such as H2NCH2CH2NH2 or N (CH2 CH2 NH2)3 or even macromolecules, such as proteins. When a ligand is bound to a metal ion through a single donor atom, as with Cl–, H2O or NH3, the ligand is said to be unidentate.
When a ligand can bind itself through two donor atoms as in H2NCH2CH2NH2 (ethane-1,2-diamine) or C2O42- (oxalate), the ligand is said to be bidentate, and when several donor atoms are present in a single ligand as in N (CH2CH2NH2)3, the ligand is said to be polydentate. Ethylenediaminetetracetate ion (EDTA4-) is an important hexadentate ligand. It can bind through two nitrogen and four oxygen atoms to a central metal ion.
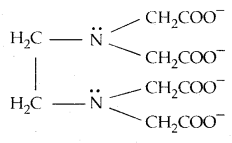
When a di- or polydentate ligand uses its two or more donor atoms to bind a single metal ion, it is said to be a chelate ligand. The number of such ligating groups is called the denticity of the ligand. Such complexes,
called chelate complexes tend to be more stable than similar complexes containing unidentate ligands. The ligand, which can ligate through two different atoms is called ambidentate ligand. Examples of such ligands are the NO2– and SCN– ions. NO2– ion can coordinate either through v nitrogen or through oxygen to a central metal atom/ion
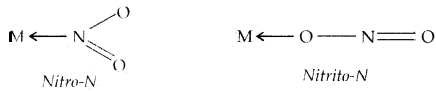
→ Coordination Number: The coordination number (CN) of a metal ion in a complex can be defined as the number of ligand donor atoms to which the metal.is directly bonded. For example, in the complexions, [PtCl6]2+ and [Ni(NH3)4]2+ the coordination number of Pt and Ni are 6 and 4 respectively. Similarly, in the complexions, [Fe(C2O4)3]2- and [Co(en)3]3+, the coordination number of Fe and Co both is 6 because C2O42- and en (ethane-1, 2-diamine) are bidentate ligands.
It is important to note here that the coordination number of the central atom/ ion is determined only by the number of sigma bonds formed by the ligand with the central atom/ion. Pi bonds, if formed between the ligand and the central atom/ion, are not counted for this purpose.
Homoleptic and Heteroleptic Complexes [Co(NH3)6]3+ in which metal is bound to only one kind of donor group, i.e., NH3 is called homoleptic complex. [Co(NH3)4 Cl2]+ complex in which a metal is bound to more than one kind of donor groups is called heteroleptic complex.
→ Nomenclature of Coordination Compounds: The formulae and names for coordination entities are based on the recommendations of the International Union of Pure and Applied Chemistry (IUPAC).
Formulae of Mononuclear Coordination Entities:
The following rules are applied while writing the formulae:
- The central atom is listed first.
- The ligands are then listed in alphabetical order. The placement of ligand in the list does not depend upon its charge.
- Polydentate ligands are also listed alphabetically. In the case of abbreviated ligand. The first letter of the abbreviation is used to determine the position of the ligand in alphabetical order.
- The formula for the entire coordination entity, whether charged or not, is enclosed in square brackets. When ligands are polyatomic, their formulae are enclosed in square brackets. When ligands are polyatomic their formulae are enclosed in parenthesis. Ligand abbreviations are also enclosed in parenthesis.
- There should be no space between the names of the ligands and the metal within a coordination sphere.
- When the formula of a charged coordination entity is to be written without that of the counter ion, the charge is indicated outside the square brackets as a right superscript with the number before the sign. For example, [Co(CN)6]3-, [Cr (H2O)6]3+ etc.
- The charge of the cations is balanced by the charge of the anions.
Note: The 2004 IUPAC draft recommends that ligands will be sorted alphabetically, irrespective of the charge.
The naming of Mononuclear Coordination Compounds Rules:
- The cation is named first.
- The ligands are named in alphabetical order before the name of the central atom/ion.
- Names of the anionic ligands end in – O. No special ending for neutral ligands .and cationic ligands. Aqua for H20, amine for NH3, carboxyl for CO and nitrosyl for NO, cyano for CN–, Oxo for O2-.
- Prefixes mono, di, tri etc. are used to indicate the number of individual ligands in the coordination entity. When the names of the ligands include a number, then the terms bis, tris takes are used, the ligand to which they refer is placed in parenthesis. For example, [NiCl2 (P Ph3)] is named as dichloro bis (triphenylphosphine) nickel (II).
- The oxidation number/state of the metal in cation, anion or neutral coordination entity is indicated by a Roman numeral in parenthesis.
- If the complexion is an anion, the name of the metal ends with -ate. For example Cr in [Cr(CN)6]3- is chromate,
- The neutral complex molecule is named the same as that of the complex cation.
Note: The 2004IUPAC draft recommends that anionic ligands will end with – ido so that chloro would become chloride, etc.
Examples:
- Cr [(NH3)3 (H2O)3]Cl3: triamine tri aqua chromium (III) chloride.
- [Co (H2N CH2CH2NH2)3]2 (SO4)3: tris (ethane -1,2 di-ammine) Cobalt (III) sulphate.
- [Cr(H2O)6] Cl3: Hexaaquachromium (111) chloride.
- [Ag(NH3)2] [Ag(CN)2]: diammine silver (I) dicyano argentate (I).
→ Isomerism in Coordination Compounds: Isomers are two or more compounds that have the same chemical formula, but different arrangements of atoms.
1. Stereoisomerism:
(a) Geometrical isomerism,
(b) Optical isomerism.
2. Structural Isomerism:
(a) Linkage isomerism,
(b) Coordination isomerism,
(c) Ionisation isomerism,
(d) Solvate isomerism.
Stereoisomers have the same chemical formula and chemical bonds but they have different special arrangements. Structural isomers have different bonds.
→ Geometrical Isomerism: It arises in heteroleptic complexes due to different possible geometric arrangements of the ligands. If in square planar complex [MX2L2], the two ligands X are on the same side and two ligands L are on the other, it is called CIS-isomer. If the two ligands X and L are opposite to each other, it is called a Trans-Isomer.
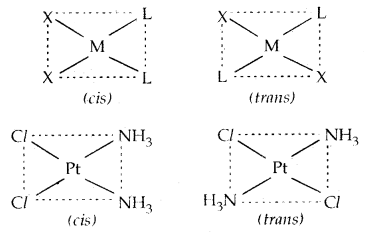
Example: [Pt (NH3)2 Cl2]: diammine dichloroplatinum (II)
Octahedral complexes [CO(NH3)4Cl2]+ and [Co{en)2Cl2]+ exist as cis and trans isomers.
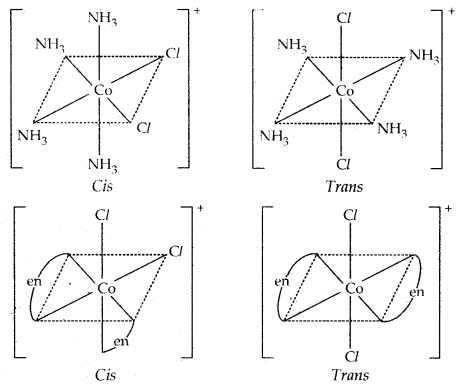
2. Optical isomerism: The isomers which rotate the plane polarised light equally but in opposite directions are called optical isomers. The isomer which rotates the plane polarised light to right is called dextrorotatory (designated as d-) while the one which rotates the plane of polarised light to the left is called laevorotatory (designated as 1). The main requirement for optical activity is that the molecule/ion should not have a plane of symmetry.
For example, complexes such as [Co(en)3]3+ and [Cr(ox)3]3- exist as optical isomers.
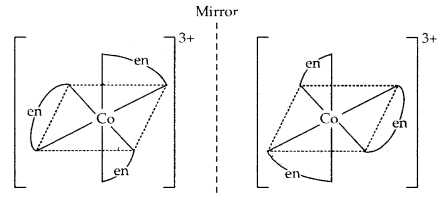
Another example of the optical isomers is shown by the complex [Co(en)2 Cl2]+.
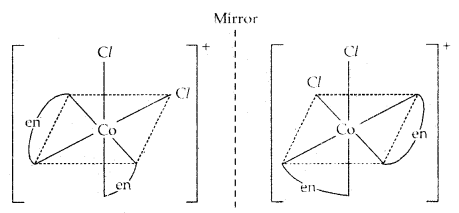
2. Structural Isomerism: They are further divided into:
1. Linkage Isomerism: It arises in a coordination compound containing ambidentate ligand. The complex [CO(NH3)5(NO2)]Cl2 exists in two forms: the red form in which the nitrite ligand is bound through oxygen (- ONO) and the yellow form in which nitrate is bound through nitrogen (-NO2). ,
2. Coordination Isomerism: It arises due to the interchange of ligands between cationic and anionic entities of different metal ions present in a complex. For example
[CO(NH3)6] [Cr (CN)6] and [Cr (NH3)6] [CO(CN)6]
3. Ionisation Isomerism: This isomerism arises when the counter ion m a complex salt is itself a potential ligand and can displace a ligand which can then become the counter ion.
(a) [CO(NH3)4O2]NO2 gives NO2– ions in solution and [CO(NH3)4 Cl(NO2)]Cl which gives Cl ions in solution.
(b) [CO(NH3)5 SO4] Br which gives a ppt. with AgNO3 [of AgBr] and [Co (NH3)Br[ SO4 which gives ppt. with Bad2 solution.
4. Solvate or Hydrate Isomerism: It is similar to ionisation isomerism with the only difference that water (H2O) is involved as a \ solvent. For example aqua complex Cr[(H2O)6]Cl3 – violet and its solvate isomer [Cr(H2O)5 Cl] Cl2. H2O – grey-green.
Bonding in Coordination Compounds:
There are two theories,
1. Valence Bond Theory for Bonding in Coordination Compounds: This theory was developed by Linus Pauling in 1930.
The basic assumptions of the theory are:
(a) The central metal atom in the complex must make available a number of empty orbitals equal to its coordination number for accommodating the electrons from ligands.
(b) The appropriate atomic orbitals (s, p, d) of the metal hybridise to give a new set of equivalent hybrid orbitals which are directed towards the ligand sites.
(c) The d orbitals used for hybridization may be either inner (n – 1)d orbitals or outer nd orbitals.
(d) The hybrid orbitals of the metal overlap with the filled orbitals of\the ligands to form coordinate bonds.
Thus, With the help of V.B. theory, the geometry of the complex can be predicted if the number of unpaired electrons is known. Alternatively, the number of unpaired electrons can be predicted from the known geometry of the complex.
The common types of geometries and hybrid orbitals used are:
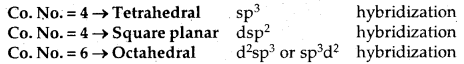
The theory may be illustrated by two important examples:
1. [CO(NH3)6]3+ and [COF6]3-. In these complexes, Co (III) has six d- electrons. The first complexion is diamagnetic and the second has paramagnetic character due to four unpaired electrons. In the [CO(NH3)6]3+ complex, the two 3d electrons get paired up with the other two leaving two vacant orbitals and these vacant orbitals get d2sp3 hybridized. In the second [COF6]3- complexes, the 3d electrons are not disturbed and the outer 4d orbitals are used for hybridization.
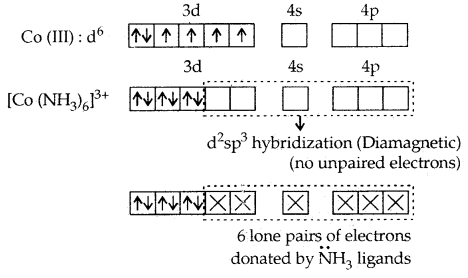
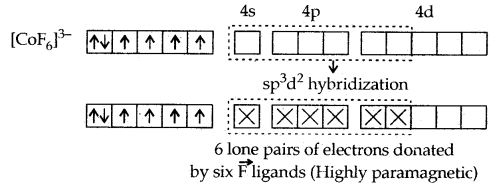
[CO(NH3)6]3+ is called an inner orbital or low spin or spin paired complex. The paramagnetic octahedral complex [COF6]3- is failed outer orbital or high spin or spin-free complex.
In a tetrahedral complex one s and three p-orbitals are hybridized to form four equivalent orbitals oriented tetrahedrally, e.g., [Ni Cl4]2-.
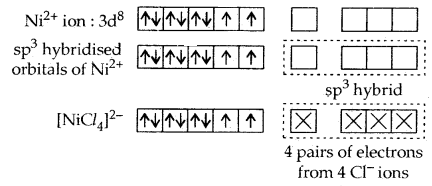
It is paramagnetic. (High spin complex).
In the square planar complexes, the hybridisation involved is dsp2. An example is [Ni(CN)4]2-. Here nickel is in a + 2 oxidation state and has the electronic configuration 3d8.
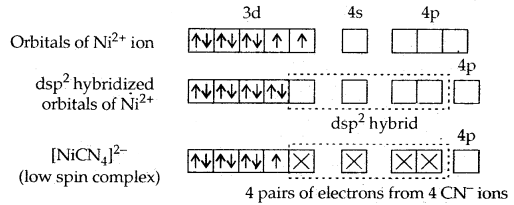
It is a diamagnetic complex.
Magnetic properties of Coordination Compounds: For metal ions with up to three electrons in the d-orbitals like Ti3+ (d); V3+ (d2); Cr3+ (d3) two vacant d orbitals are available for octahedral hybridisation with 4s and 4p orbitals. The magnetic behaviour of these free ions and complex is similar.
When more than three 3d electrons are present, the required pair of 3d orbitals for octahedral hybridisation is not directly available (as a consequence of Hund’s rule). Thus, for d4 (Cr2+, Mn3+), d5 (Mn2+, Fe3+), d6 (Fe2+, CO3+) cases, a vacant pair of d orbitals results only by the pairing of 3d electrons which leaves two, one and zero unpaired electrons respectively.
The magnetic data agree with maximum spin pairing in many cases, especially with coordination compounds containing d6 ions. However, with species containing d4 and d5 ions, there are complications. [Mn (CN)6]3- has a magnetic moment of two unpaired electrons while [MnCl6]3- has a paramagnetic moment of four unpaired electrons. [Fe(CN)6]3- has a magnetic moment of a single unpaired electron while [FeF6]3- has a paramagnetic moment of five unpaired electrons. [COF6]3- is paramagnetic with four unpaired electrons while [Co(C2O4)3]3- is diamagnetic.
This apparent anomaly is explained by valence bond theory in terms of the formation of inner orbital and outer orbital coordination entities. [Mn(CN)6]3-, [Fe(CN)6]3- and [Co(C2O4)3]3- are inner orbital complexes involving d2 sp3 hybridisation, the former two complexes are paramagnetic and the latter diamagnetic. On the other hand, [MnCl]3-/ [FeF6]3- and [COF6]3- are outer orbital complexes involving sp3d2 hybridisation and are paramagnetic corresponding to four, five and four unpaired electrons.
Limitations of Valence Bond Theory:
- It involves a no. of assumptions.
- It does not give a quantitative interpretation of magnetic data.
- It does not explain the colour shown by coordination compounds.
- It does not explain the thermodynamic or kinetic stabilities of co-ordinate compounds.
- It does not make exact predictions regarding the tetrahedral and square planar structures of 4-coordination complexes.
- It does not distinguish between weak and strong ligands.
2. Crystal Field Theory: This theory envisages the metal ligand to be purely ionic arising.from electrostatic interactions between the metal and ligand. Ligands are treated as point charges in case of anions or dipoles in case of neutral molecules. The five d orbitals in an isolated gaseous metal atom/ion have the same energy, i.e., they are degenerate.
On the arrival of the ligands, these d-orbitals split up. Those orbitals which lie on the direct path of the ligands are repelled more than those which lie away from the path of approaching ligands. This pattern of splitting depends upon the nature of the crystal field.
A. Crystal Field splitting into Octahedral Coordination Entities: In an octahedral complex when the six ligands approach the metal atom/ ion, out of five d-degenerate orbitals, three-d orbitals (dxy, dyz, dzx) which lie away from the path of the approaching ligands (∵ they lie in between the axes) are repelled less than those two d-orbitals (dx2–y2 and dz2) which lie on the direct path [they lie in between the axes].
Thus the degeneracy of the five d-orbitals are lost and these d-orbitals split up. This splitting gives two sets of orbitals -12 set of three orbitals of lower energy and eg set of two orbitals (dx2–y2 and dz2) of higher energy. It is called Crystal Field Splitting and the difference of energy is denoted by Δ0 (o -for octahedral complex). The energy of two eg orbitals is raised by \(\frac{3}{5}\) Δ0 and that of the three t2g will decrease by \(\frac{2}{5}\) Δ0.
The crystal field splitting, Δ0, depends upon the field strength of the ligands which is in the order:
I– < Br < SCN– < Cl– < S2- < F– < OH– < C2O42- < H2O < NCS– < edta4- < NH3 < en < CN– < CO such a series is called spectrochemical series.
In d1 complexes, the single d-electron occupies the lower t2g orbital. In d2 and d3 coordination entities, the d-electrons occupy the three t2g orbitals singly in keeping with Hund’s Rule.
For d4 ions, two different patterns are possible:
- the 4th electron could either enter the lower energy t2g level and pair with an existing electron, or
- it could occupy the higher energy e2g level. It depends upon two factors:
(a) The magnitude of crystal field splitting energy Δ0.
(b) The pairing energy P [energy required for electron pairing in a single orbital).
1. If Δ0 < P, the 4th electron enters one of the eg orbitals. Ligands for which Δ0 < P is known as Weak field ligands and form high spin complexes.
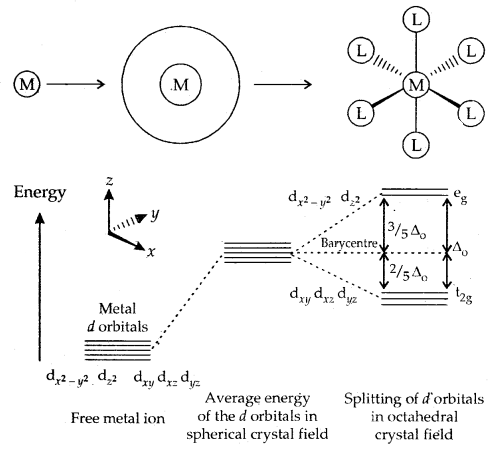
Crystal Field Splitting of d-orbitals in an Octahedral complex
2. If Δ0 > P, the 4th electron occupies a lower energy t2g orbital. Ligands producing this effect are known as strong field ligands and form low spin complexes.
Calculations show that d4 to d7 coordination entities are more stable for the strong field as compared to weak field cases.
B. Crystal Field Splitting into Tetrahedral Coordination Entities: In tetrahedral complexes, splitting of d-orbitals is such that it is opposite to that of octahedral complexes. The eg set of d-orbitals of the metal ion is lower in energy and the t2g set has higher energy. Moreover, the value of Δt [t for tetrahedral complex] is smaller than Δ0 (Δt = \(\frac{4}{9}\) Δ0) for the same ligands and metal-ligand distances. Consequently, the orbital splitting energies are not sufficiently large for forcing pairing of electrons, i.e., electrons prefer to remain unpaired and thus low spin complexes are rarely observed in tetrahedral complexes.
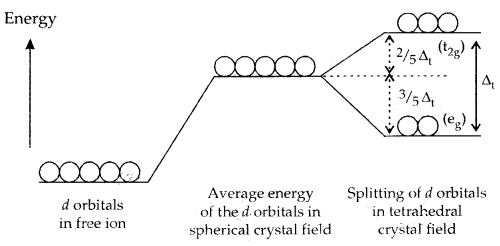
d-orbitals splitting in tetrahedral crystal field
Colour in Coordination Compounds: Formation of coloured complexes is .the characteristic property of transition elements. It can be explained readily on the basis of crystal field theory, taking an example of an octahedral complex of [Ti (H2O)6]3+ in which the metal ion Ti3+ is a 3d1 system.
The t2g set is lower in energy and the eg set is higher in energy. The rotary 3d1 electron prefers to remain in t2g set in the ground state. If the light corresponding to a yellow-green region of white light is absorbed by this complex, it will excite the electron from the t2g set to the next available eg
set[t2g eg° → t°2g eg1 ]. Thus the complex appears violet.
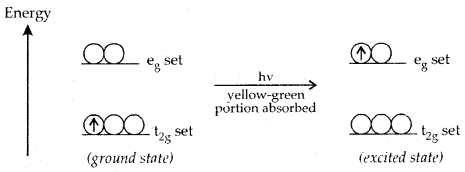
The transition of an electron in [Ti(H2O)6]3+ complex
The colour of the complexes is explained by crystal field theory due to the d-d transition of the electron. Removal of the ligands does not cause crystal field splitting and hence the complex becomes colourless,
e.g., where CuSO4.5H20 [Complex: [Cu (H2O)4]2+. SO42-. H2O] is blue in colour due to absorption of the red region of white light, anhydrous CuSO4 s white. Removal of water from [Ti(H2O)6]Cl3 on heating renders it colourless.
The influence of the ligand on the colour of a complex may be illustrated by considering the [Ni (H2O)6]2+ complex, which forms when nickel (II) chloride is dissolved in water. If the identity ligand, ethane- 1, 2-diamine (en) is progressively added in the molar ratios en: Ni, 1:1, 2:1,3:1 the following series of reactions with associated colour changes occur.
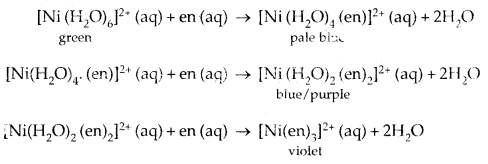
Limitations of Crystal Field Theory:
- As ligands are assumed to be point charges, anionic ligands are expected to have a greater splitting effect. However, actually, they are found to be at the lower end of the spectrochemical series.
- It does not take into account the covalent character of bonding between the ligand and the central atom/ion.
- Though OH– ion in the spectrochemical series lies below H2O and NH3, yet it produces a greater splitting effect.
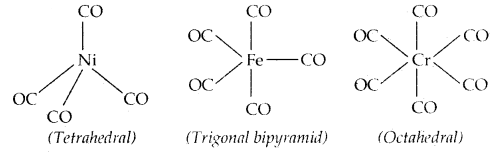
→ Bonding in Metal Carbonyls: The homoleptic carbonyls (compounds containing carbonyl ligands only) are formed by most of the transition metals. Tetracarbonylnickel (o), viz., [Ni (CO)4] is tetrahedral, Penta carbonyl iron (o) is trigonal bipyramidal while Hexa carbonyl chromium (o) is octahedral.
Stability of Coordination Compounds: Consider the reaction
M + 4 L → ML4
Stability constant (on Equilibrium constant)
K = \(\frac{\left[\mathrm{ML}_{4}\right]}{\left[\mathrm{M} \mid[\mathrm{L}]^{4}\right.}\)
The numerical value of the stability constant is a measure of the stability of the complex in the solution.
Importance And Applications of Coordination Compounds: These compounds are widely present in the mineral, plant and animal world. They play important functions in analytical chemistry, metallurgy, biological systems, industry and medicine.
1. Hardness of water is estimated by simple titration with Na2 EDTA. The Ca2+ and Mg2+ ions present in hard water make stable complexes with EDTA.
2. Metallurgical extraction of silver and gold make use of complex formations. Gold can be extracted from [Au (CN)2]– complex by the addition of zinc.
3. Purification of Nickel can be achieved by Mond’s process by converting impure nickel to complex [Ni (CO)4] which decomposes to yield pure nickel.
4. Chlorophyll, the green pigment present in plants responsible for photosynthesis, is a coordination compound of Magnesium. Haemoglobin, the red blood pigment of blood that acts as an oxygen carrier is a coordination compound of iron. Vitamin B12, Cyanocobalamin, the anti-pernicious anaemia factor, is a cord in. m compound of cobalt.
5. Coordination comp us are used as catalysts for many industrial processes. Rhodium complex [(Ph3P) RhCl]. a Wilkinson catalyst is used for the hydrogenation of alkenes.
6. Articles can be electroplated with silver and gold much more smoothly and evenly from a solution of complexes Ag (CN)2]– and [Au (CN)2]– than from a solution of simple metal ions.
7. In black and white photography, the developed film is fixed by washing with hypo solution which dissolves the under-composed AgBr to form a complex io,n [ Ag (S2O3)2]3-.
8. Cis-platin is used for the treatment of cancer. Excess of copper and iron are removed by the chelating ligands D-penicillamine and desferrioxamine B via the formation of coordination compounds. EDTA is used in the treatment of lead poisoning