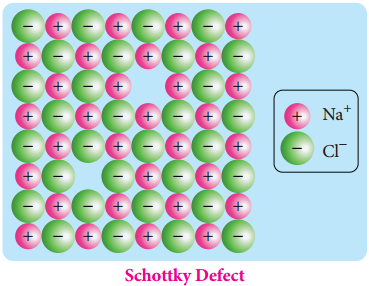
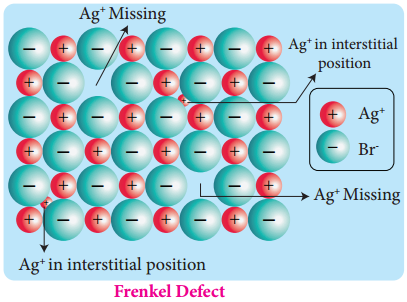
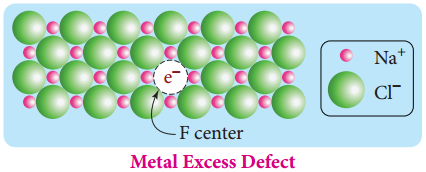
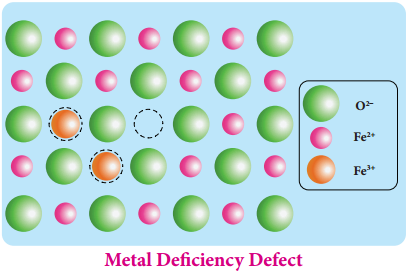
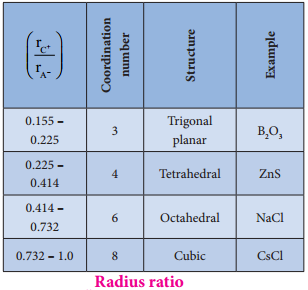
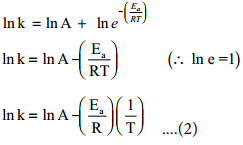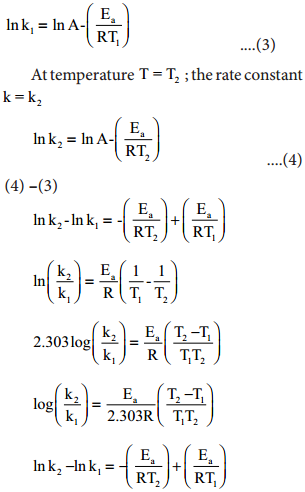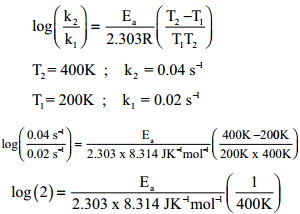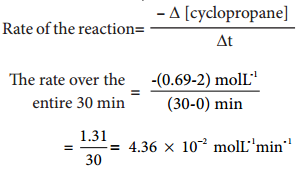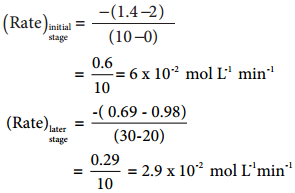Find free online Chemistry Topics covering a broad range of concepts from research institutes around the world.
Factors Affecting the Reaction Rate
The rate of a reaction is affected by the following factors.
- Nature and State of the Reactant
- Concentration of the Reactant
- Surface Area of the Reactant
- Temperature of the Reaction
- Presence of a Catalyst
Nature and State of the Reactant:
We know that a chemical reaction involves breaking of certain existing bonds of the reactant and forming new bonds which lead to the product. The net energy involved in this process is dependent on the nature of the reactant and hence the rates are different for different reactants.
Let us compare the following two reactions that you carried out in volumetric analysis.
- Redox reaction between ferrous ammonium sulphate (FAS) and KMnO4
- Redox reaction between oxalic acid and KMnO4
The oxidation of oxalate ion by KMnO4 is relatively slow compared to the reaction between KMnO4 and Fe2+. In fact heating is required between KMnO4 and Oxolate ion and is carried out at around 60°C.
The physical state of the reactant also plays an important role to influence the rate of reactions. Gas phase reactions are faster as compared to the reactions involving solid or liquid reactants. For example, reaction of sodium metal with iodine vapours is faster than the reaction between solid sodium and solid iodine.
Let us consider another example that you carried out in inorganic qualitative analysis of lead salts. If you mix the aqueous solution of colorless potassium iodide with the colorless solution of lead nitrate, precipitation of yellow lead iodide take place instantaneously, whereas if you mix the solid lead nitrate with solid potassium iodide, yellow coloration will appear slowly.

Concentration of the Reactants:
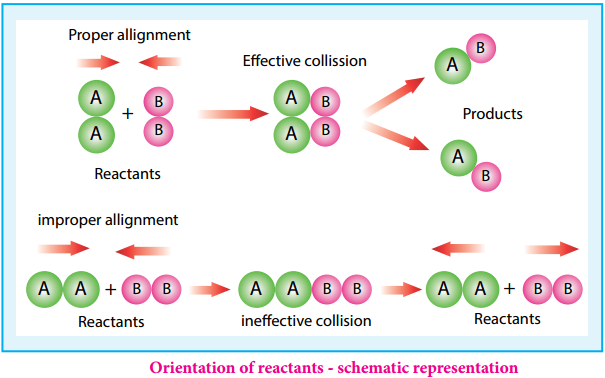
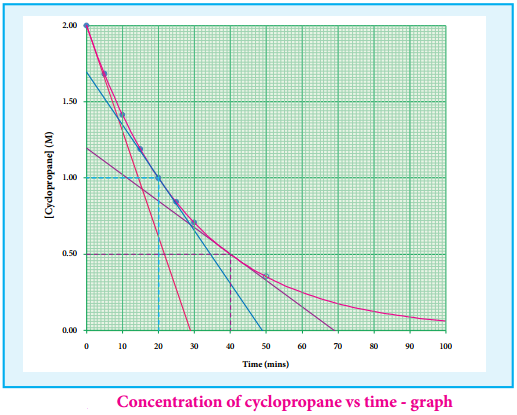
The rate of a reaction increases with the increase in the concentration of the reactants. The effect of concentration is explained on the basis of collision theory of reaction rates. According to this theory, the rate of a reaction depends upon the number of collisions between the reacting molecules. Higher the concentration, greater is the possibility for collision and hence the rate.
Effect of Surface Area of the Reactant:
In heterogeneous reactions, the surface areas of the solid reactants play an important role in deciding the rate. For a given mass of a reactant, when the particle size decreases surface area increases. Increase in surface area of reactant leads to more collisions per litre per second, and hence the rate of reaction is increased. For example, powdered calcium carbonate reacts much faster with dilute HCl than with the same mass of CaCO3 as marble.
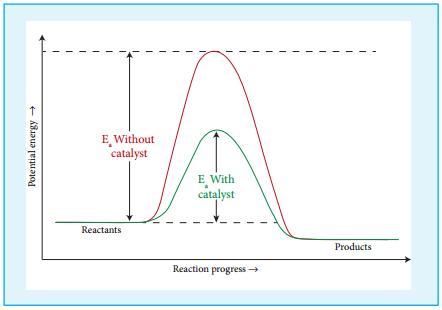
Effect of Presence of Catalyst:
So far we have learnt, that rate of reaction can be increased to certain extent by increasing the concentration, temperature and surface area of the reactant. However significant changes in the reaction can be brought out by the addition of a substance called catalyst.
A catalyst is substance which alters the rate of a reaction without itself undergoing any permanent chemical change. They may participate in the reaction, but again regenerated at the end of the reaction. In the presence of a catalyst, the energy of activation is lowered and hence, greater number of molecules can cross the energy barrier and change over to products, thereby increasing the rate of the reaction.